

Diagnóstico infrecuente de pubertad detenida
Infrequent diagnosis of arrested puberty
Caso Clínico
Dras. Zunana C1, Peña, TM2, Mattone MC3, Costanzo M4, Guercio G5, Califano P6.
Resumen
Las consultas y preocupaciones sobre el desarrollo puberal son frecuentes en la atención de niñas y adolescentes. Se presenta el caso de una adolescente femenina de 14 años con pubertad detenida e hipogonadismo hipergonadotrófico, cuya etiología fue desarrollo sexual diferente por alteración del desarrollo gonadal 46,XY. En el seguimiento, desarrolla disgerminoma y gonadoblastoma. Se discute el abordaje complejo e interdisciplinario de estos casos. El tratamiento implica considerar aspectos relacionados a la terapia de reemplazo hormonal y el tratamiento de las gónadas en función del riesgo de malignización, entre otros.
Palabras claves: pubertad detenida, hipogonadismo hipergonadótrofico, desarrollo sexual diferente tumores de células germinales, gonadectomía.
Abstract
Complaints and concerns about pubertal development are frequent in the care of girls and adolescents. We present the case of a 14-year-old female adolescent with arrested puberty and hypergonadotrophic hypogonadism due to different sexual development secondary to 46,XY disordered gonadal development. During follow-up, dysgerminoma and gonadoblastoma were developed. The complex and interdisciplinary approach is discussed. Treatment should address issues related to hormone replacement therapy, and gonadal management as in function of gonadal malignancy risk, among others.
Keywords: arrested puberty, hypergonadotrophic hypogonadism, differences in sexual development, germ cell tumors, gonadectomy.
Introducción
Las consultas y preocupaciones sobre el desarrollo puberal son motivos de consulta frecuentes en la atención de niñas y adolescentes. La pubertad detenida se define como la falta de progresión puberal durante más de 2 años después de su inicio espontáneo.1 Esta definición, según otros autores, es considerada como amenorrea primaria.2
La falta de progreso puberal puede presentarse como:
1) la ausencia de la menarca 2 años después desde el inicio de la Telarca;
2) desarrollo deficiente de las mamas; y
3) un crecimiento de talla subóptima.
La pubertad detenida es considerada patológica hasta que se demuestre lo contrario y debe evaluarse con urgencia.1
Para definir la causa, se deberán estudiar alteraciones centrales que afecten al eje hipotalámico-pituitario (hipogonadismos hipogonadotróficos funcionales u orgánicos) y alteraciones de las gónadas (hipogonadismo hipergonadotrófico). Una detallada anamnesis, examen físico exhaustivo y estudios de laboratorio podrán identificar la mayor parte de las etiologías. En la evaluación inicial de estas adolescentes, es conveniente, en primera instancia, descartar embarazo. Si la anatomía genital fuera típica con vagina permeable y útero, se sugiere realizar un perfil gonadotrófico.2
El objetivo de esta publicación es presentar las dificultades diagnósticas y de seguimiento de una adolescente mujer con pubertad detenida.
Caso Clínico
Adolescente mujer de 14 años y 2 meses que comienza seguimiento en este hospital, derivada por Pediatría para evaluación puberal con sospecha de pubertad detenida. Refería telarca a los 11 años. En la anamnesis, no refiere antecedentes médicos o quirúrgicos de importancia. En la entrevista a solas, se autopercibe como mujer cisgénero. Concurre al colegio, no realiza actividad física fuera del establecimiento. Tiene amigos, no está en pareja, tiene orientación heterosexual y no ha iniciado relaciones sexuales.
Al examen físico, se encuentra eutrófica. Presenta como índices antropométricos 58.7 Kg (percentil 75), talla: 150 cm (percentil 10), índice de masa corporal 26.1 kg/m2. Se evidencian mamas y vello pubiano en estadio de Tanner 3 e introito con dos orificios; la vagina es permeable y está estrogenizada. Axilarca positiva. No presenta signos de hiperandrogenismo clínico. No se palpan formaciones inguinales. Adjunta ecografía de otro centro con útero de 4 cm de longitud y ovarios pequeños de 0,5 y 0,6 cc. Edad ósea de 11 años, según método de Greulich & Pyle.
Los estudios de laboratorio informan: FSH 93UI/L, LH 29.6UI/L, E2 30pg/mL, 17OHP 0.3ng/mL, testosterona < 0.12ng/ml, sulfato de dehidroepiandrosterona 480ng/ml, TSH 1.38 µU/ml y prolactina 9.4ng/ml. Una segunda determinación (FSH 94.2 UI/L, LH 25UI/L, E2 35 pg/mL) confirma el diagnóstico de hipogonadismo hipergonadotrófico. Se solicita cariotipo en sangre periférica, cuyo resultado es 46,XY. Esto establece el diagnóstico de desarrollo sexual diferente (DSD) por alteración del desarrollo gonadal 46,XY (disgenesia gonadal completa 46,XY).
Se indica realizar valoración interdisciplinaria y se explica la necesidad de exploración gonadal por el riesgo de tumor de células germinales asociado a esta entidad. Asimismo, se explica posibilidad de comenzar con terapia de reemplazo hormonal.
El abordaje de la paciente y su familia resulta dificultoso debido a la negación en recibir la información y realizar controles, lo que determina la pérdida de seguimiento durante, aproximadamente, un año. Al retomar, cuando ya la paciente cuenta con 15 años y 6 meses de edad, se realiza ecografía que informa en región anexial derecha y se detecta la presencia de una formación redondeada con calcificaciones con sombra acústica que mide 3.5x2.2x1.7cm con escaso parénquima periférico conservado (Figura 1 y 2). Los marcadores tumorales subunidad βhCG, LDH, CA 125, CEA, CA 19.9 y Alfa feto proteína resultan negativos.
Se indica realizar exploración laparoscópica, a la cual tanto la adolescente como su familia se encuentran reticentes. Se discute el caso en el Comité de Desarrollo Sexual Diferente integrado por los siguientes servicios: social, salud mental, endocrinología, neonatología, bioética, asuntos jurídicos, cirugía, urología, ginecología y pediatría. Se trabajan estrategias médicas, psicológicas y legales para abordaje. Luego de múltiples entrevistas, acceden al procedimiento. Los estudios moleculares en genes candidatos para el diagnóstico de DSD disponibles no evidenciaron alteraciones.
Durante el procedimiento quirúrgico, se constata gónada izquierda que impresiona cintilla gónada (streak) y gónada derecha con formación sólida de 4cm de diámetro de aspecto neoplásico, por lo que se realiza anexectomía bilateral. La anatomía patológica informa presencia de disgerminoma de gónada derecha con tumor limitado a la misma que mide 3,5 x 2,3 x 2 cm y gonadoblastoma. La gónada remanente (muy escasa) es compatible con cintilla clásica con área de hiperplasia de células de Leydig (aproximadamente 3 mm). Con dichos resultados, es evaluada por oncología, quien realiza la estadificación. Se realizan tomografías de cerebro y tórax, centellograma óseo corporal total, resonancia magnética de abdomen y pelvis con contraste endovenoso, sin evidenciar lesiones. La paciente se estadifica en estadio P1 posquirúrgico por el Comité de tumores, por lo cual se recomienda realizar seguimiento estricto con imágenes y marcadores tumorales séricos según protocolos vigentes. La familia y la adolescente aceptan y entienden la importancia de controles estrictos.
A los 6 meses se decide, en conjunto con la adolescente, iniciar terapia hormonal de reemplazo con valerato de estradiol por vía oral en dosis crecientes, que toma erráticamente.
A lo largo del seguimiento se trabajan, con mucha dificultad, distintos aspectos del diagnóstico. En la entrevista a solas, se discuten, con la paciente, pautas de salud sexual. No desea hablar sobre el embarazo o fertilidad. Refiere escolaridad acorde, tiene proyectos a futuro. Niega consumos problemáticos de sustancias. Niega ideas autolesivas o situaciones de violencia sexual. Se explica la importancia de uso de barrera para la prevención de infecciones de trasmisión sexual, así no menstrúe.
A los 18 años y medio, y luego de 6 meses de recibir dosis de estrógeno adultas (2 mg de valerato de estradiol) con Tanner 4 y útero puberal con 0,9 cm de endometrio, se rota a anticonceptivos hormonales con etinilestradiol de 0.03 mg y levonorgestrel de 15 mcg en rango extendido, ya que no deseaba deprivar. Concurre a los 6 meses del inicio de anticonceptivos; refiere buena tolerancia y deprivación controlada durante el descanso.
Se discuten con la adolescente distintos aspectos médicos y de información antes de su transición, que se realiza en forma exitosa a un centro de adultos especializado.
Discusión
El término “desarrollo sexual diferente/intersex” o “DSD”, por sus siglas en inglés (disorders/differences of sex development), se utiliza para definir aquellas condiciones congénitas en las cuales el desarrollo del sexo cromosómico, gonadal o anatómico es atípico. Su incidencia varía entre 5/1000 recién nacidos a 1/4500, según las condiciones consideradas. Su etiología es muy diversa, así como su forma de presentación, y el abordaje suele ser un desafío para el equipo de salud.3
La clasificación de DSD más utilizada es la descripta en el Consenso Chicago de 2005. Propone dividir las etiologías en función del cariotipo cromosómico (desbalance afectando al par de cromosomas sexuales); 46,XX y 46,XY (Tabla 1).3
En los casos como el presentado, la constitución cromosómica es “masculina” (XY), pero el fenotipo es “femenino”. Estas situaciones pueden producirse por afectación de la diferenciación testicular o por compromiso en la síntesis o acción de las hormonas involucradas en la diferenciación sexual (andrógenos). La alteración del desarrollo gonadal testicular o disgenesia testicular puede ser completa o parcial. En las formas completas, como en la adolescente descripta, el desarrollo testicular es prácticamente nulo, lo que se refleja en la falta de producción de la testosterona necesaria para la virilización de los genitales internos y externos por las células de Leydig, y la falta de síntesis y secreción de la hormona antimülleriana (AMH) por las células de Sertoli. Esto se traduce en la presencia de un fenotipo femenino con la presencia de derivados de Müller. En estas situaciones, el laboratorio presenta niveles de testosterona y AMH bajos en relación al cariotipo 46,XY. Se sugiere realizar una imagen inicial (ecografía) para evaluar las estructuras müllerianas, la localización gonadal y su ecoestructura. El estudio se completa con la biopsia gonadal y el estudio molecular de genes de diferenciación gonadal (SRY, WT1, etc.).4
Dentro del grupo de los DSD 46,XY, la mayoría suele diagnosticarse en la infancia por la presencia de genitales atípicos. Sólo un pequeño grupo se diagnostica en la adolescencia, sobre todo aquellos casos con fenotipo femenino completo. Los motivos de consulta en estos casos suelen ser retraso puberal, pubertad detenida, amenorrea primaria o algún signo de virilización (hirsutismo o clitoromegalia).5
En la evaluación de cualquier adolescente que consulta por retraso puberal, pubertad detenida o amenorrea primaria, con estudios de laboratorio que evidencian hipogonadismo hipergonadotrófico, sin antecedentes que justifiquen el daño gonadal, es mandatorio solicitar un cariotipo en sangre periférica. El cariotipo 46,XY con presencia de derivados müllerianos y sin valores elevados de andrógenos sugiere el diagnóstico de DSD 46,XY por disgenesia gonadal. Entre un 10 y 20% de estos casos se asocia a variantes que afectan al gen SRY. Lamentablemente, en un 50 % de estos casos, aun con las técnicas más modernas de investigación, la etiología final permanece desconocida.4
En relación con desarrollo mamario presentado por la adolescente descripta, se ha postulado que los niveles normales de estrógeno en casos con disgenesia gonadal podrían atribuirse hipotéticamente a varios mecanismos, entre ellos: la producción de estrógenos por las gónadas streak en el momento de la pubertad, la conversión periférica de andrógenos en estrógenos y el aumento de la sensibilidad del tejido mamario a los estrógenos. Además, se ha informado que tanto los esteroides sexuales masculinos como femeninos pueden haber sido producidos por el gonadoblastoma. Este tumor es, a menudo, la fuente de hormonas en las mujeres con el cariotipo 46,XY. Algunos autores ya han descripto desarrollo sexual mamario en adolescentes con cariotipo 46,XY y gonadoblastoma en gónadas disgenéticas. Además, se han informado otros casos con menstruación espontánea debido a la secreción de estrógenos neoplásicos.
El abordaje debe ser interdisciplinario, incluyendo el manejo de la información, el sostén psicoemocional y los eventuales requerimientos de terapia hormonal y cirugía. 6
La comunicación de la información debe realizarse con lenguaje claro, con términos exactos y acorde a las posibilidades y los tiempos para la compresión de la adolescente y su familia.3,5,7 Cuando existan intereses contrapuestos entre la familia y la adolescente, deberá primar el interés superior del niño y el derecho a la información. No obstante, la información del diagnóstico final no es una urgencia y es recomendable trabajarla con la adolescente y la familia, paulatinamente. El trabajo de la información debe ser realizado por equipos con experiencia para evitar mensajes discordantes y uso de terminología peyorativa, confusa y heteronormativa. Se necesitan explicar los conceptos básicos de genética, desarrollo sexual y acción de las hormonas, y aclarar que la identidad es un fenómeno de autopercepción y no está determinado por ningún factor cromosómico o anatómico. Suele ser muy útil el apoyo en gráficos. Se ha propuesto que el intercambio de experiencias con grupos de pares con igual o similar patología pueden ser beneficioso.7
En una adolescente como la descripta en este trabajo, con disgenesia gonadal completa y fenotipo femenino, la conducta quirúrgica se limita al manejo de las gónadas. La gonadectomía profiláctica es una decisión compleja que debe individualizarse considerando: género asignado al nacer, el riesgo de malignización, su función (tanto hormonal como su potencial fertilidad) y la posibilidad de vigilancia.
Según publicaciones recientes sobre gonadectomía profiláctica en las disgenesias gonadales 46,XY, el riesgo de desarrollo de tumores de células germinales es intermedio-alto (varía del 12 al 60 %), dependiendo de la alteración molecular. En el escenario de esta paciente, se recomienda la gonadectomía bilateral precoz. La evaluación histológica e inmunohistoquímica minuciosa de las gónadas es fundamental para la identificación de lesiones premalignas y malignas. 8,9
Es importante que la adolescente comprenda la importancia del tratamiento hormonal sustitutivo tanto en corto como largo plazo. No existe un acuerdo generalizado acerca del mejor momento para iniciarla, las dosis iniciales ni tampoco su ritmo de aumento. La mayoría de los grupos destacan la necesidad de iniciar el tratamiento farmacológico con una dosis baja y continuar con un aumento progresivo, pero con diferencias en cuanto a la edad del inicio y a la dosificación en los primeros años.10
Otro aspecto que nunca debe descuidarse es la consejería en salud sexual y reproductiva. Esta debe realizarse con perspectiva de género, con foco en una sexualidad placentera y segura, con protección para infecciones de transmisión sexual. En relación con la posibilidad de maternar, se pueden plantear las técnicas de reproducción asistida con ovodonación (en caso de desarrollo adecuado de las estructuras müllerianas) y la adopción.11
Consideraciones finales
La pubertad detenida, en raras oportunidades, puede deberse a disgenesia gonadal. En aquellos casos donde el diagnóstico es DSD 46,XY, el abordaje es complejo y requiere de un trabajo interdisciplinario. La devolución diagnóstica a la adolescente y su familia es un proceso que debe respetar los tiempos de cada binomio. El tratamiento debe abordar cuestiones relacionadas a la terapia de reemplazo hormonal, la gonadectomía profiláctica o terapéutica en relación con el riesgo de malignización gonadal y la consejería en salud sexual y reproductiva.
Anexo
Figura 1. Ecografía ginecológica transabdominal.
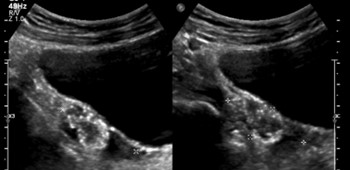
Gónada derecha con formación redondeada con calcificaciones de 3.6 x2.8 x 2 cm que reemplaza casi por completo la estructura de la misma.
Figura 2. Ecografía ginecológica transabdominal.
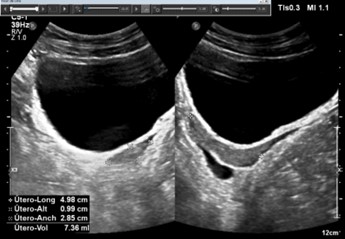
Útero en anteversoflexión de 4,89 cm x 0,99 por 2,85 cm sin diferenciación endometrial. Escaso liquido libre en fondo de saco de Douglas.
Tabla 1. Clasificación DSD.
|
Cromosómico |
DSD 46,XY |
DSD 46,XX |
|
● 45,X y variantes ● 47,XXY y variantes ● 45,X/46,XY (mosaicismos) ● 46,XX/46,XY (quimera/ ovotesticular) |
Desorden del desarrollo gonadal -DSD ovotesticular Desordenes de la biosíntesis o acción de los andrógenos Malformaciones Hipogonadismo hipogonadotrófico |
Desorden del desarrollo gonadal Exceso de andrógenos de origen extragonadal -Hiperplasia suprarrenal congénita (21OHD) -Otros déficits enzimáticos (ej.: -Andrógenos maternos -Otros: malformaciones |
Adaptado de Bailez et al, “Desarrollo sexual diferente”.3
Bibliografía
[1] Wei, Christina et al. “The investigation of children and adolescents with abnormalities of pubertal timing.” Annals of clinical biochemistry vol. 54,1 (2017): 20-32.
2 Klein, David A et al. “Amenorrhea: A Systematic Approach to Diagnosis and Management.” American family physician vol. 100,1 (2019): 39-48.
3 Bailez, Marcela et al. “Desarrollo sexual diferente”. Programa Nacional de Actualización Pediátrica 2018. Sociedad Argentina de Pediatría. Módulo 1. Capítulo 1. Páginas: 17-47.
⁴ Guerrero-Fernández, Julio et al. “Guía de actuación en las anomalías de la diferenciación sexual (ADS) / desarrollo sexual diferente (DSD)” [Management guidelinesfordisorders / different sex development (DSD)]. Anales de pediatria vol. 89,5 (2018): 315.e1-315.e19.
5 Davies, Kate. “The XY Female: Exploring Care for Adolescent Girls with Complete Androgen Insensitivity Syndrome.” Comprehensive child and adolescent nursing vol. 43,4 (2020): 378-388.
6 Çatlı, Gönül et al. “An Unusual Presentation of 46,XY Pure Gonadal Dysgenesis: Spontaneous Breast Development and Menstruation.” Journal of clinical research in pediatric endocrinology vol. 7,2 (2015): 159-62
7 Baratz, Arlene B et al. “Disorders of sex development peer support.” Endocrine development vol. 27 (2014): 99-112.)
8 Cools, Martine et al. “Managing the risk of germ cell tumourigenesis in disorders of sex development patients.” Endocrine development vol. 27 (2014): 185-96.
9 Guerrero-Fernández, Julio et al. “Consensus guide on prophylactic gonadectomy in different sex development.” Endocrinología, diabetes y nutrición vol. 69,8 (2022): 629-645.
10 Hiort, Olaf. “Long-term management of patients with disorders of sex development (DSD).” Annales d'endocrinologie vol. 75,2 (2014): 64-6.6
11 Guercio G, Costanzo M, Grinspon RP, Rey RA. Fertility Issues in Disorders of Sex Development. Endocrinol Metab Clin North Am. 2015 Dec;44(4):867-81.
1,2. Médica asistente de Ginecología Infantojuvenil.
3,6. Becaria del Servicio de Endocrinología.
4,6. Jefa de Cínica Médica Endocrinología.
5. Jefa de Clínica Ginecología Infantojuvenil.
Hospital Nacional de Pediatría “Prof. Dr. Juan P Garrahan”.
6. Consejo Nacional de Investigaciones Científicas y Técnicas. CONICET.