

Retrasos en la maduración puberal: reflexiones sobre dos casos clínicos
Delayed puberty: consideration about two clinical cases
Caso Clínico
Dra. Metella Dei (¹), Dra. Chiara Immacolata Meleca (²)
Resumen
El diagnóstico y tratamiento de adolescentes con retraso de la maduración puberal puede seguir siendo un desafío para los pediatras, endocrinólogos y ginecólogos, porque el mismo cuadro clínico puede ser determinado por causas muy diferentes. La aparición de la menarquia, como etapa final del desarrollo puberal requiere tanto de la integridad anatómica del eje hipotálamo-hipófisis-ovario y del tracto útero-vaginal como de un normal funcionamiento de los distintos genes reguladores de los sistemas endocrinos involucrados y de una adecuada disponibilidad energética y de homeostasis metabólica.
En este artículo enfocamos las diferencias entre los déficit funcionales de la hormona liberadora de gonadotropina hipotalámica (GnRH) (hipogonadismos hipogonadotrópicos) no asociados con otras patologías, y los retrasos puberales constitucionales.
Actualmente no disponemos de marcadores bioquímicos específicos capaces de diferenciar con precisión entre estas dos situaciones. Utilizaremos dos casos clínicos como temas de reflexión: el primero definido al final del proceso diagnóstico como hipogonadismo hipogonadotrópico relacionado con mutaciones genéticas del receptor del GnRH y el segundo como un retraso puberal, a pesar de la presencia de una anomalía estructural de la región hipofisaria.
Los conocimientos genéticos y clínicos actuales, también apoyados por algunos estudios longitudinales, sugieren que consideremos una visión unitaria de estos cuadros clínicos. Esta idea plantea una serie de interrogantes, por ejemplo sobre el momento adecuado para iniciar cualquier terapia de inducción puberal fisiológica, siguiendo los métodos de administración y posología propuestos por las guías internacionales.
Palabras clave: Retraso puberal. Hipogonadismo hipogonadotrópico. Sistema hipotalámico-hipofisario. Genética de la maduración puberal. Terapia de inducción puberal.
Abstract
The diagnostic work-up and the management of adolescents with pubertal delay is sometimes a challenge for pediatricians, endocrinologists, and gynecologists, because the same clinical picture can be the result of extremely different causes. The appearance of the menarche, as final milestone of pubertal development, requires in fact the anatomical integrity of hypothalamus-pituitary-ovarian axis and of the genital tract, as well as a normal function of genes regulating the endocrine systems involved together with an adequate energy availability and metabolic homeostasis.
In the present article, we particularly focus the differences between functional deficiencies of Hypothalamic Gonadotropin Releasing Hormone (GnRH), i.e. hypogonadotropic hypogonadisms not related to other pathologies and constitutional delays of puberty. Currently, there are no specific biochemical markers to accurately differentiate these two disorders. We will describe use two case reports as topics for reflection. The first has been defined at the end of the diagnostic work-up as hypogonadotropic hypogonadism linked to genetic mutations of GnRH receptor; the second as pubertal delay in spite of the presence of a structural anomaly of the pituitary region.
Current genetic and clinical knowledges, supported by few longitudinal studies, suggest considering a unified vision of these clinical pictures. These leanings pose some questions, for instance about the right time to start an eventual therapy of physiological pubertal induction, following the methods of administration and the dosages proposed by international guidelines.
Key words: pubertal delay, hypogonadotropic hypogonadisms, hypothalamic-pituitary axis, genetics of pubertal maturation, pubertal induction therapy
Introducción
Clínicamente, el retraso puberal se define como la ausencia de menarca a los 15 años con presencia de caracteres sexuales secundarios, la ausencia completa de desarrollo mamario a los 13 años o la no aparición de la menarquia 4 años después de la telarca. Estos criterios se construyen considerando una edad promedio de menarquia poco después de los 12 años y un intervalo promedio entre telarca y menarca de 2 años y medio. Por lo tanto, deben adaptarse a poblaciones en las que los estándares de referencia son diferentes.
En muchos casos, el retraso puberal representa una variante retardada de una maduración puberal fisiológica (el llamado retraso constitucional del crecimiento y de la pubertad), pero este es un diagnóstico de exclusión no simple, ya que una gran variedad de condiciones clínicas pueden determinar un desarrollo puberal retrasado o incluso ausente (Tabla 1).
En la Figura 2 reportamos la frecuencia de las más importantes patogénesis de retraso puberal relativa a los casos de la Unità di Ginecologia Infanzia e Adolescenza de la Università di Firenze, Italia. Se observa que, excluyendo los cuadros secundarios de insuficiencia ovárica que son prevalentes, las formas de hipogonadismo funcional (principalmente trastornos alimenticios) y los retrasos puberales constitucionales, representan las formas clínicas más comunes.
El diagnóstico diferencial a veces también puede incluir cuadros de falla del desarrollo del tracto genital inferior (hipoplasia mülleriana o síndrome de Rokitansky-Kunster-Hauser-Meyer o, más raramente, malformaciones obstructivas) que, sin embargo, son identificables con la consulta médica y con una evaluación ecográfica. Las formas relacionadas con la falla ovárica son evidentes con un perfil endocrino básico, para niveles elevados de FSH. El estudio endocrino siempre puede fácilmente excluir otras patologías (hiperprolactinemia, hipotiroidismo, etc) capaces de interferir en la reactivación fisiológica del eje GnRH-gonadotropinas.
Los cuadros de hipogonadismo llamado funcional, es decir secundario a situaciones de estrés psicofísico, déficit energético por actividad física excesiva o trastornos alimenticios restrictivos han ido aumentando en los últimos años. Y es el clima endocrino del comienzo de la maduración puberal que tiende a acentuar tanto la predisposición genética que la presión ambiental sobre la preocupación por el peso y la imagen corporal ([1]) No siempre la anamnesis, los datos antropométricos (crecimiento de estatura, IMC) o las dosis hormonales de primer nivel (gonadotropinas, estradiol, hormonas tiroideas) nos dirigen al diagnóstico.
A menudo, es necesario para comprender realmente la situación que subyace proponer a la adolescente un diario alimenticio o una monitorización del gasto energético de la actividad física, junto a una evaluación de la composición corporal (análisis de impedancia bioeléctrica) ([2]) y otros indicadores endocrinos de estado metabólico (FT3, Insulina, IGF-1, cortisol).
Pero aún más difícil es a menudo discriminar entre un retraso puberal constitucional y un déficit de gonadotropinas aislado, es decir no asociado con otros signos, enfermedades (como la celiaquía) o déficit (incluida la hiposmia): en este punto focalizaremos nuestra discusión. Quizás sean precisamente nuestras herramientas diagnósticas que aún no son capaces de definir completamente estos cuadros clínicos que, solo en ocasiones se aclaran con un seguimiento en el tiempo. Por lo tanto nos vamos a detener sobre las características y las posibilidades diagnósticas de esas formas que llegan a nuestra observación sin otros síntomas o signos de importancia, con una desaceleración más o menos pronunciada de la progresión puberal. Con la ayuda de dos casos clínicos, también discutiremos el significado de algunas herramientas de diagnóstico.
¿Déficit puberal aislado o retraso en la maduración?
- Patologías orgánicas hipotalámo-hipofisarias potencialmente silentes
Si consideramos las patologías orgánicas, tanto congénitas como adquiridas, que pueden influir en las etapas de maduración puberal (Tabla 2), muchos cuadros son diagnosticables en base a la anamnesis, signos o síntomas neurológicos o perfil endocrino. Sin embargo, existen algunas alteraciones morfofuncionales del sistema hipotálamo-hipofisario que se evidencian únicamente por la resonancia magnética (RM) con medio de contraste de gadolinio. Esto explica por qué esta evaluación, aunque costosa y desagradable para la paciente joven, entró en el proceso de diagnóstico de estos cuadros clínicos.
Puede ser la causa de retraso puberal una hidrocefalia obstructiva que causa una estenosis del acueducto de Silvio que determina la dilatación del tercer ventrículo, que se localiza en el diencéfalo en la región suprahipotalámica. Puede ser una patología de base genética o adquirida, por ejemplo, por compresión por parte de un quiste aracnoideo. La distensión de la región peri-ventricular y medio-basal del hipotálamo determina la interrupción de la secreción de GnRH. Algunas veces puede asociarse también a hipotiroidismo y anomalías de la secreción de GH, pero con mayor frecuencia no son evidentes otros déficits endocrinos y no implican necesariamente cefalea o síntomas neurológicos, siendo la curva de crecimiento y el desarrollo de caracteres sexuales secundarios suele ser normal. Los niveles de LH y estradiol están dentro de lo normal o en los límites inferiores del rango normal para la edad. Por lo tanto, el diagnóstico también puede ser una confirmación a través de RM sin ninguna sospecha clínica más que el comienzo tardío de la menarquia ([3]).
La silla vacía es una alteración anatómica caracterizada por la presencia de líquido en el interior de la silla turca con compresión de la adenohipófisis en el suelo selar. En la adolescencia suele ser una forma primaria, es decir, no relacionada con patologías o lesiones conocidas. Es un cuadro frecuente que, en ocasiones se diagnostica por un hallazgo accidental, por ser completamente asintomática. Puede asociarse también con diversas disfunciones endocrinas, incluido el déficit de GH, la hiperprolactinemia o el déficit de gonadotropinas primaria o secundaria ([4]). Por tanto, se puede presentar como una amenorrea primaria con perfil endocrino normal.
Las hipofisitis son cuadros flogísticos heterogéneos que afectan a la glándula pituitaria; entre estos, el más específico es la forma autoinmune o linfocitaria, que es consecuencia de un proceso de destrucción de las células hipofisarias secretoras por los linfocitos T reactivos. Se caracteriza a nivel histológico por una infiltración de linfocitos, macrófagos y células plasmáticas. Ocurre con mayor frecuencia durante el embarazo y el posparto, pero se han descrito varios casos en edad pediátrica y en la adolescencia. La hipofisitis a veces se presenta en pacientes con diátesis autoinmunitaria, con enfermedad relacionada con Ig G4 o es secundaria al uso de fármacos biológicos inmunomoduladores que activan las células T. Es asociada a antígenos de histocompatibilidad HLA DQ8 y DR53 y pueden estar presentes anticuerpos anti-pituitaria (APA) y en ocasiones también antihipotálamo (AHA). Puede manifestarse con cefalea y aumento de peso. La hiperprolactinemia puede estar presente aunque en general hay un déficit gonadotrofico ([5]). En la RM se presenta con un engrosamiento del tallo hipofisario y un aumento del volumen de la glándula que puede ser similar a un adenoma hipofisario. El diagnóstico diferencial no es fácil ([6]); se cree que la prevalencia real es mayor de los cuadros diagnosticados.
- Hipogonadismos hipogonadotrópicos aislados
Se estima una prevalencia de 1:7000/8000, con una relación hombre/mujer: 3.6. Son formas cuya amplia heterogeneidad genética ha sido intensamente estudiada en los últimos años; esto también ha implicado una profundización de nuestro conocimiento sobre la fisiología del desarrollo intrauterino de las neuronas secretoras de GnRH y su regulación.
Los estudios genéticos han ayudado a comprender cómo existe una interacción compleja entre genes que en la vida intrauterina orientan la migración de neuronas secretoras de GnRH, regulan las neuronas olfativas que actúan como su guía y guían las proyecciones de los distintos axones; así como en el período posnatal y en la maduración puberal, múltiples genes activan los mecanismos de regulación de las neuronas secretoras de GnRH, los objetivos periféricos de su producción hormonal y la función de las gonadotropinas. Nos remitimos a la literatura específica para obtener más información ([7];[8]), subrayando lo importante que es tener una anamnesis familiar precisa sobre los tiempos de maduración puberal y sobre cualquier terapia realizada para promover la maduración sexual.
Clínicamente, las adolescentes con hipogonadismo hipogonadotrópico tienen un crecimiento normal, por lo tanto, una tasa de crecimiento > 3.6 cm/año, sin una aceleración evidente del crecimiento puberal. El desarrollo mamario es deficiente, aunque variable; la adrenarquia está a menudo presente, pero se describen también situaciones de vello púbico limitado en las primeras etapas puberales. El estudio ecográfico de los genitales internos revela un déficit de crecimiento uterino y ovarios con presencia de microfolículos, pero sin signos de maduración folicular. La edad ósea tiende a retrasarse. Las concentraciones de LH, FSH, inhibina B están en los límites inferiores del rango, pero no son por si mismos diagnósticos. Los niveles de E₂ son bajos, a veces apenas detectables en relación con la sensibilidad del método de análisis. La hormona antimülleriana (AMH) suele ser normal, lo que indica una reserva ovárica preservada [9];[10];[11] Respecto a los exámenes de segundo nivel, la respuesta a la prueba de estímulo con GnRH no suele confirmar el diagnóstico, aunque los datos de la literatura se refieren principalmente a casos masculinos[12]. La resonancia magnética del encéfalo y del tronco del encéfalo por medio de contraste se indica siempre para excluir anomalías del eje hipotálamo-hipofisario silentes y para evaluar los lóbulos olfativos; también se están estudiando otros parámetros diagnósticos, como el volumen hipofisario, cuyo crecimiento está relacionado con la producción de estrógenos y hormonas de la adrenarca[13]; [14]
- Retrasos puberales constitucionales
Tienen una prevalencia de alrededor del 2% en las adolescentes. En el 50-80% de los casos hay familiaridad; en las jóvenes predomina la herencia por línea materna, con una transmisión del 50 al 80% de los casos es autosómica dominante, pero con penetrancia incompleta[15] . También se describen modos de transmisión oligogénicos, es decir que involucran variantes de más de un gen[16]. Incluso en el caso de retrasos constitucionales, se demuestra una clara heterogeneidad genética que involucra mutaciones de genes que regulan la migración de neuronas a GnRH, de los sistemas que modulan la secreción de GnRH incluyendo el sistema de kisspeptina - neuroquinina B - dinorfina, los receptores de GnRH, FSH, GH y genes implicados en la homeostasis metabólica[17].
Incluso a nivel clínico, los retrasos puberales son heterogéneos. Por un lado, tenemos pacientes con talla normal al nacer y posterior desaceleración del crecimiento que se inicia en los primeros dos o tres años de vida, resultando una estatura entre el 3° y 10° percentil al comienzo de la pubertad que se asocia a una ralentización de la maduración puberal: el llamado retraso constitucional del crecimiento y desarrollo (RCCD). Por otro lado tenemos a los individuos con estatura por debajo del 50° percentil, pero de acuerdo con el retraso en el desarrollo de los caracteres sexuales secundarios: el retraso puberal constitucional (RPC). El intervalo entre telarca, pico de crecimiento y menarquia progresa con algunos años de retraso, las etapas de la pubertad fisiológica. La edad ósea tiende a retrasarse por la edad cronológica, pero de acuerdo con la altura y el estadio de Tanner. Las concentraciones de FSH, LH, estradiol, IGF-1, hormonas tiroideas también están tendencialmente de acuerdo con la maduración puberal y la edad ósea. Los niveles de inhibina B se están estudiando como marcador de diagnóstico, pero los datos publicados son predominantemente para hombres[18];[19] Incluso el uso de pruebas de estímulo no permite un esclarecimiento diagnóstico sin un seguimiento en el tiempo; también se incluye el estudio de la respuesta a la kisspeptina que aún es una propuesta experimental[20].
Caso clínico 1
Giovanna acude a una primera consulta a los 14 años y 8 meses, enviada por el pediatra de la familia, por sospecha de alteración puberal. Ha iniciado la telarca bilateral desde hace más de dos años, pero se ha mantenido casi sin cambios durante el último año. La pubarca también está presente. La radiografía de la mano y muñeca izquierda para la edad ósea (método de Greulich & Pyle) mostró una edad ósea de 12 años y 6 meses.
Anamnesis familiar: la madre tuvo menarca poco después de los 11 años y sufre de endometriosis pélvica. El padre tuvo un desarrollo puberal aparentemente normal. Sufre de migrañas. La hermana mayor con menarca a los 12 años tiene ciclos regulares y padece tiroiditis de Hashimoto. La prima (hija de la hermana de la madre), en cambio, tuvo un retraso en la pubertad con menarca a los 15 años y 11 meses; ahora tiene 18 años y tiene ciclos regulares. Abuelo materno con diabetes tipo 2 que comenzó alrededor de los 50 años. Abuela materna con cáncer mamario posmenopáusico. No consanguinidad. No familiaridad de episodios tromboembólicos.
Anamnesis personal: nacida a término, con peso al nacer: 3,110 kg. Lactancia materna durante 3 meses. Asma alérgica especialmente en la niñez con uso ocasional de corticosteroides inhalantes, suspendido desde hace algunos años. No alergias a medicamentos. No toma otros medicamentos. Crecimiento estatural alrededor del 50° percentil (sin pico evidente de crecimiento estatural). Dentición en tiempos fisiológicos. Refiere una dieta no selectiva (confirmada por la madre). Patina dos horas a la semana. Ella informa un sentido del olfato aparentemente normal (le gustan mucho los perfumes). Tiene buenos resultados académicos.
Examen físico: no revela dismorfismo; no presenta acné ni seborrea. El índice de masa corporal (IMC) es 18.8. Normotensa. La evaluación según Tanner: Estadio III de mama y vello pubiano; no hay vello en las axilas. Los genitales externos son normales, con algunos signos de estrogenización. La ecografía pélvica transabdominal muestra un cuerpo uterino medio de 4 cm de largo, con una relación cuerpo/cuello: 1.1. El borde endometrial es apenas visible. Ambos ovarios tienen un volumen de 0.5 cm3 con evidentes microfolículos pequeños.
Exámenes de laboratorio:
FSH: 4.3 UI/l - LH: 1 mU/ml - PRL: 2.75 mcg/l (2-25) - E2: 27 pg/ml
inhibina B: 4.2 pg/ml (11 aa < 4.8 pg/ml) - 17OH progesterona: 1.07 mcg/l - cortisol: 14.8 mcg/dl DHEAS: 303 mcg/dl (28-320) - TSH: 2.35 mU/ml - FT3: 3.75 ng/l (2.7 -5.7) - FT4: 0.83 ng/dl (0.7 -1.7) glucemia: 78 mg/dl - Insulina: 10.45 mcU/ml - IGF-1: 216.1 ng/ml –
anticuerpos (IgA) anti transglutaminas: 4 U/ml (< 7U/ml) - IgA totales: 230 mg/dl (90-400 mg/dl) creatinemia: 0.90 mg/dl
Dados los bajos niveles de LH se realiza resonancia magnética (RM) encéfalo y tronco encefálico con medio de contraste (bajo profilaxis con corticoides) cuyo informe revela que no hay alteraciones de señal focal o diseminación del parénquima cerebral y cerebeloso; no áreas de restricción de la propagación del medio de contraste. Cuerpo calloso normal según la morfología, grosor y señal. Adenohipófisis de tamaño normal en sitio. Tallo hipofisario de volumen en los límites inferiores para la edad. Neurohipófisis normalmente representada. Aspecto quístico de la pineal. Cavidades ventriculares normales por localización, morfología, volumen y señal. Ancho de los espacios del LCR peri-cerebral y la permeabilidad de las cisternas de la base normales. Estructuras de línea central en eje.
La evaluación morfológica de la región hipotalámica-hipofisaria en este caso no es de particular ayuda diagnóstica: de hecho, la estandarización de la medida del pedículo todavía está en estudio y especialmente no está claramente correlacionada con la clínica[21].
Al no estar claro aún el diagnóstico diferencial entre el retraso puberal constitucional o el hipogonadismo hipogonadotrópico, se propone un control dentro seis meses para evaluar la progresión de los caracteres sexuales secundarios con el control de FSH, LH, E2, la inhibina B y pruebas de coagulación en la hipótesis de iniciar un tratamiento más tarde. Mientras tanto, su pediatra de referencia insiste en realizar también un GnRH, que muestra una adecuada respuesta en LH al estímulo (Tabla 3).
Consulta de control: Giovanna tiene 15 años y 3 meses. No ha tenido problemas de salud. Ha continuado con una dieta regular, manteniendo inalterada su actividad física. Está preocupada por la falta de menstruación y porque sus senos no han cambiado. Ha crecido 2 cm de altura y 1 kg de peso. Actualmente tiene un índice de masa corporal (IMC): 18.75.
En el examen clínico muestra un estadio de Tanner T3 P3 sin cambios. La ecografía pélvica transabdominal muestra un cuerpo uterino medio de 4 cm de largo, con una relación cuerpo/cuello: 1.2. El borde de endometrio es apenas perceptible. El ovario derecho tiene un volumen de 0.6 cm3; el ovario izquierdo de 0.5 cm3; en ambos hay varios microfolículos con un diámetro máximo de 5 mm. Las pruebas endocrinas muestran:
FSH: 3.9 mUI/ml - LH: 1.2 mU/ml - E2 < 25 pg/ml - inhibina B: 3.8 pg/mL (rango para 11 a < 4.8 pg/ml) - Proteína C anticoagulante, proteína S, aPCR, AT III normales.
Se decide iniciar una terapia hormonal de reemplazo con estradiol transdérmico semanal, empezando los primeros dos meses con una dosis de 12.5 цg y después de dos meses de 25 цg, planificando una prueba ecográfica al final del cuarto mes y una densitometría lumbar.
En la hipótesis de hipogonadismo hipogonadotrópico, se remite a asesoramiento genético.
Al comienzo del cuarto mes de tratamiento (por debajo de 25 цg E2) se verifica un manchado prolongado y, ataques de migraña. Giovanna, sin embargo, está muy feliz con sus senos que finalmente han crecido. La ecografía transabdominal realizada unos días después muestra un útero de morfología adulta con un endometrio de 6 mm. La densitometría ósea muestra una densidad mineral (DMO) en el nivel L1-L4 de 0.809 g/cm2 correspondiente a un Z-score: -2.1.
Se propone aumentar la terapia con un parche semanal de 35 цg de E2 continuando con didrogesterona doce días al mes y llevar un diario de la cefalea. Luego se verifica si la dosis de estrógeno es adecuada mediante dosificación del estradiol plasmático junto con el seguimiento en el tiempo de la tendencia de la migraña y la densidad mineral ósea.
La dosis estrogénica deberá incrementarse posteriormente, de acuerdo con las guías internacionales[22].
Pasados unos meses llega la respuesta de la asesoría genética: Cariotipo 46, XX. El análisis de genes implicados en el síndrome de Kallmann sobre ADN de sangre periférica muestra la presencia de las siguientes variantes heterocigotas:
- Gen GNRHR: c.784C>T, p.Arg262Trp en el exón 3;
- Gen GNRHR: c.317A>G, p.Gln106Arg en el exón 1 (reportado en sujetos con hipogonadismo hipogonadotrópico sin anosmia con herencia recesiva);
- Gen CHDT: c.307T>A, p.Ser103Thr en el exón 2; (mutación del gen de la acetiltransferasa de colina, probablemente no significativa).
Las configuraciones genéticas confirman el diagnóstico de hipogonadismo hipogonadotrópico por mutaciones del receptor por el GnRH. Esta es la primera alteración genética históricamente identificada en sujetos con hipogonadismo hipogonadotrópico y se refiere al 40% de los casos familiares de hipogonadismo hipogonadotrópico sin anosmia (con trasmisión autosómica recesiva) y el 17% de los idiopáticos[23].
Las mutaciones estudiadas inactivan el receptor o su capacidad de unión condicionando fenotipos variables de déficit parciales a completos de actividad gonadotrópica[24]. Se describen también mutaciones que dan retraso puberal o amenorrea secundaria. En pacientes con hipogonadismo es común un desarrollo mamario parcial, con detención o progresión puberal[25]. En la mayor parte de los casos estudiados la respuesta al GnRH exógeno es normal[26] y esto explica el resultado de la prueba de GnRH, pero también es un elemento importante en el plan terapéutico para permitir el embarazo.
La historia de Giovanna confirma que en algún hipogonadismo es posible tener una cierta maduración de los caracteres sexuales secundarios y que es sobre todo la falta de progresión en el tiempo lo que actúa como alarma. También nos muestra que el perfil endocrino tanto basal como bajo estímulo, al menos con los métodos de laboratorio actualmente en uso, no nos ofrece un diagnóstico cierto.
Caso clínico 2
Gaia tiene 14 años y medio y acude a la consulta debido a la ausencia de caracteres sexuales secundarios.
Anamnesis familiar: madre con bocio multinodular, abuela materna con neoplasia mamaria, abuela paterna con esclerodermia. El padre informa de un retraso puberal. No enfermedades cardiovasculares familiares.
Anamnesis personal: nacida de un embarazo a término con nacimiento espontáneo. Tardíamente cambió sus últimos dientes deciduos. Sufre de cefalea frontal. Recientemente tuvo algunos episodios de dolores recurrentes en miembros superiores e inferiores por lo que fue sometida a una consulta reumatológica que no encontró patologías en curso. No informa dificultades en la percepción de olores. No realiza actividades físicas de una manera estructurada y se alimenta cualitativamente «mal» dicho de su madre y de ella misma. No tiene problemas particulares en la escuela y en el grupo de amigos. Pero me dice que está bastante preocupada por la falta de desarrollo de los senos. Había realizado el año anterior pruebas para celiaquía con resultados negativos y un RX mano y muñeca izquierda para valoración de la edad ósea que estaba de acuerdo con la edad cronológica (método de Greulich y Pyle). Su curva de crecimiento muestra una tendencia estable en el 15° percentil, con una tasa de crecimiento de alrededor de 5 cm por año. Su estatura objetivo es 166,5 ± 8.
Examen físico: no muestra rasgos dismórficos o marcas distintivas. El IMC es: 22.48. La presión arterial es normal. La valoración según Tanner es: T1 (con mínima adipomastia) P2; los genitales externos no muestran signos de estrogenización.
La ecografía transabdominal muestra un cuerpo uterino de 3,8 cm de largo con una relación cuello/cuerpo < 1 y morfología infantil. Se observa alguna estructura microfolicular en ambos ovarios.
Exámenes de laboratorio:
FSH: 1.3UI/l - LH< 0.2 UI/l (método ICMA) - PRL: 4.87 mcg/l - E2: < 20 ng/l
inhibina B: 17 pg/ml - I GF-1: 105.3 mcg/l (rango de edad: 103-524)
TSH: 2.4 mUI/l - FT4. 0.92 ng/dl - FT3: 3.48 ng/l
ACTH: 15 ng/l, DHEAS: 69 mcg/dl - Cortisol: 7.9 mcg/dl - 17OHP: 1.2 ng/ml
Anticuerpos (IgA) anti transglutaminas: 5 U/ml (< 7U/ml) - IgA totales: 190 mg/dl (90-400 mg/dl)
Creatinemia: 0.8 mg/dl
Resonancia magnética de la región y tronco encefálico con medio de contraste: No se observan alteraciones de señales focales o difusas del parénquima cerebral y cerebeloso. El cuerpo calloso es normal según la morfología, volumen y señal. Neurohipófisis ectópica que se localiza en la raíz del pedúnculo; adenohipófisis en su lugar pero de volumen relativamente pequeño para la edad. Aspecto quístico de la pineal. Cavidades ventriculares normales en sitio, morfología, volumen y señal. Regular el ancho de los espacios del líquido cefalorraquídeo (LCR) peri-cerebral y la permeabilidad de las cisternas de la base. Estructuras de línea central en eje.
La ectopia de la neurohipófisis puede ser causada por la interrupción del sistema portal peri-infundibular, que habitualmente también altera la función de la adenohipófisis al reducir la llegada directa de factores reguladores hipotalámicos. La persistencia de axones tróficos neurosecretores induce la formación de una neurohipófisis ectópica, pero generalmente funcional. Se describen cuadros tanto de base genética como adquirida con informes también de posible aparición en la edad adulta. La ectopia de la neurohipófisis con hipoplasia de la adenohipófisis puede implicar también déficit de GH [27]; [28], retraso puberal o pubertad precoz central, diabetes insípida y déficits múltiples de secreción de la adenohipófisis[29]; [30]; [31].
Reevaluando la situación clínica de Gaia propongo un control de la diuresis diaria (cuidando de tomar al menos 1 litro y medio por día), una consulta nutricional para mejorar un poco el estilo de alimentación y un nuevo dosaje de LH, E2, DHEAS, IGF-1 a los 3 meses del anterior, sugiriendo nueva consulta para valorar el posible inicio de una terapia hormonal.
Consulta de control: el examen físico muestra un Tanner T2 P2. Gaia me dice que ha establecido una buena relación con la dietista que la está siguiendo y que está cuidando más su nutrición. La diuresis diaria es 1600 ml, por lo tanto normal. Los controles hormonales muestran:
LH: 2.8 UI/l - E2, 52 pg/ml - DHEAS: 75 цg/dl - IGF-1: 267.2 цg/l
Se decide no iniciar la terapia por la aparición de un verdadera telarquia y los niveles de LH y estradiol nos muestran una inicial secreción de gonadotropinas, correspondiente a un comienzo de maduración puberal. Gaia está decepcionada y no convencida de mis explicaciones, porque hubiera preferido comenzar el tratamiento de inmediato.
Luego de un año, la madre de Gaia comenta que su hija en realidad tiene senos bien desarrollados, vello púbico extenso y ha crecido 8 cm de altura. Sin embargo, mientras tanto, ha tenido algunos problemas importantes del estado de ánimo por los que es atendida por una neuropsiquiatra.
Al año siguiente Gaia concurre a una consulta: tuvo la menarquia el mes anterior (a los 16 años y 10 meses) y ya ha tenido el segundo ciclo y esto la hace feliz. Ahora mide 166 cm de altura y tiene un IMC de 21.5. También está mejor psicológicamente.
La ecografía transabdominal revela un útero mediano en anteversoflexión con morfología adulta, de 7 cm de largo, con endometrio compatible con la fase proliferativa. El ovario izquierdo tiene un volumen de 7 cm3 con evidentes formaciones foliculares en diversas etapas de maduración. El ovario derecho tiene un volumen de 6 cm3 con varios microfolículos y un folículo de 7 cm de diámetro. En esa consulta se comienza a hablar de anticoncepción.
En la historia de Gaia, la progresión de caracteres sexuales secundarios, incluida la aceleración del crecimiento, comenzó tarde, a pesar de la presencia de una anomalía importante de la región pituitaria. Nótese también la normalidad de la edad ósea ante un retraso significativo en la maduración: los datos de la literatura coinciden de hecho en un retraso en la maduración ósea (es decir, con una desviación de la edad cronológica de 2DS) especialmente en cuadros de retraso constitucional en el crecimiento y la pubertad; esto no se corresponde exactamente con la situación clínica de Gaia[32].
Conclusiones
Al evaluar a las adolescentes con retraso en el desarrollo puberal, con o sin retraso en el crecimiento, tal vez un cambio de perspectiva puede ayudarnos. En lugar de intentar plantear lo antes posible un diagnóstico diferencial entre formas evolutivas y déficit de secreción de gonadotropinas puede ser más correcto considerar los retrasos puberales constitucionales, los hipogonadismos hipogonadotrópicos y también el déficit de GH parciales como cuadros del mismo espectro de anomalías endocrinas (Figura 2).
Contamos con varios elementos que validan este paradigma:
Los estudios genéticos han demostrado una gran superposición entre estas formas17, en el sentido de que diferentes mutaciones de los mismos genes dan lugar a diferentes fenotipos. Esto también se demuestra por los hallazgos de la anamnesis: por ejemplo, el 10% de los familiares de los sujetos con hipogonadismos hipogonadotrópicos tiene un retraso puberal constitucional[33]. De hecho, conocemos mejor las mutaciones que corresponden a situaciones de déficit de gonadotropinas, pero se están estudiando incluso las que corresponden a los retrasos puberales y esto permitirá en un futuro, tener un panel de pruebas que pueden ayudar en diagnosis diferencial[34].
Incluso algunos estudios clínicos confirman la visión de una interconexión entre los diversos cuadros clínicos. Por ejemplo, con respecto a la hiposmia, que es un síntoma clave en el hipogonadismo del síndrome de Kallmann, se ha demostrado, con pruebas precisas, un amplio espectro de variaciones en la capacidad olfativa en muchos sujetos con hipogonadismo hipogonadotrópico, sin anomalías evidentes de los bulbos olfativos correspondientes a mutaciones de los distintos genes implicados (KAL1, PROKR2, FG8, FGFR1, GnRH, TAC3)[35];[36].
También en el nivel de la función menstrual, la diferencia entre retrasos constitucionales e hipogonadismos no está clara. El hipogonadismo hipogonadotrópico por definición implica una amenorrea primaria, pero en 8-9% de los casos se ha informado algún sangrado menstrual incluso en ausencia de terapia de reemplazo hormonal, en particular en portadoras de déficit del receptor de GnRH [37]:[38]. Además, especialmente en sujetos varones, se ha documentado una reversibilidad en el tiempo de algunas formas de hipogonadismo hipogonadotrópico, tanto en un estudio prospectivo que la resalta en 10% de los casos[39], que en estudios retrospectivos que documentan una posibilidad de activación funcional del 5 al 14% de los casos [40]; [41]; [42]; [43]. También hay informes en los sujetos femeninos, con varias mutaciones documentadas, de reversibilidad a largo plazo, tal vez después de un período de terapia de reemplazo hormonal[44];[45]; 42.
Estas consideraciones tienen algunas implicaciones a nivel clínico:
- en caso de duda diagnóstica, es importante explicar claramente la situación y la necesidad de seguir a lo largo del tiempo la evolución del cuadro clínico para aclarar la patología subyacente;
- dado que la falta de signos de maduración puberal es un tema que puede desencadenar reacciones emocionales profundas con riesgo de aislamiento, una vez completado el proceso diagnóstico, es posible, aun cuando estemos orientados hacia un diagnóstico de retraso puberal constitucional, proceder con un ciclo corto de terapia con estradiol, preferiblemente transdérmica o percutáneo[46], dosis bajas durante seis meses con posterior evaluación. Sin embargo, es importante individualizar la dosis según el estadio mamario alcanzado la etapa puberal mamaria alcanzada y, el peso corporal de acuerdo con las instrucciones para una inducción puberal fisiológica[47] para evitar cambios rápidos y, a menudo, no favorables a un armónico desarrollo mamario e interferencias en el crecimiento estatural.
Por lo tanto se utilizan, por lo general, parches de transdérmicos que disponen de 25 цg dividiéndolos en 2 partes, ya que ha estado documentada la posibilidad de almacenar las partes cortadas en ≤ + 35° C[48]. Sin embargo no tenemos estudios longitudinales relativos a pacientes femeninas que nos muestren que en caso de retraso puberal constitucional la terapia de reemplazo es beneficiosa en términos de crecimiento estatural y especialmente la mejora de la masa ósea, cuya reducción representa la repercusión a distancia más importante de la maduración puberal tardía[49].
Finalmente, son necesarios más investigaciones longitudinales con estudios genéticos y que comparen los efectos de las terapias apropiadas con los de un comportamiento de espera con respecto a variables físicas (altura final, alcanzar el pico de masa ósea, fertilidad) y psicológicas.
Tablas
Tabla 1: Condiciones clínicas que se pueden manifestar con retraso puberal
|
Nivel de déficit |
Cuadro clínico |
Patologías |
|
Patología malformativa del tracto genital inferior. |
Ausencia de menarquia con caracteres sexuales secundarios normales. Dolor abdominal-pélvico en presencia de obstrucción con endometrio funcional. |
Malformaciones obstructivas cervicovaginales Hipoplasia mülleriana o síndrome de Rokitansky Kunster Hauser Meyer |
|
Déficit gonadal |
Ausencia de menarquia con desarrollo variable de caracteres sexuales secundarios. |
Disgenesia gonadal Déficit gonadal adquirida |
|
Déficit gonadotrópico |
Ausencia de menarquia con desarrollo tendencialmente lento de caracteres sexuales secundarios. |
Patologías orgánicas del sistema nervioso central Hipogonadismo hipogonadotrópico de base genética Retrasos constitucionales en el crecimiento y la pubertad. Hiperprolactinemia Endocrinopatías con repercusión en el crecimiento (déficit de Gh, hipotiroidismo, Cushing ...) Síndrome de Ovario Poliquístico Hipogonadismo funcional (déficit energético, celiaquia, Crohn, otras enfermedades crónicas) Exposición a contaminantes endocrinos Formas sindrómicas (Charge, Prader Willi, Noonan, síndrome cubital-mamario...) |
Tabla 2: Patologías orgánicas del sistema nervioso central que pueden provocar retraso o déficit puberal
|
Tabla 3 Prueba de GnRH: respuesta de FSH y LH a 100 mcg de GnRH por vía intravenosa
|
Tiempos |
FSH (mUI/ml) |
LH (mUI/ml) |
|
0 ' |
3,25 |
1,27 |
|
15 ' |
7.02 |
12.28 |
|
30 ' |
6,91 |
12.30 |
|
60 ' |
8,37 |
10,36 |
|
90 ' |
7.53 |
7.13 |
Figuras
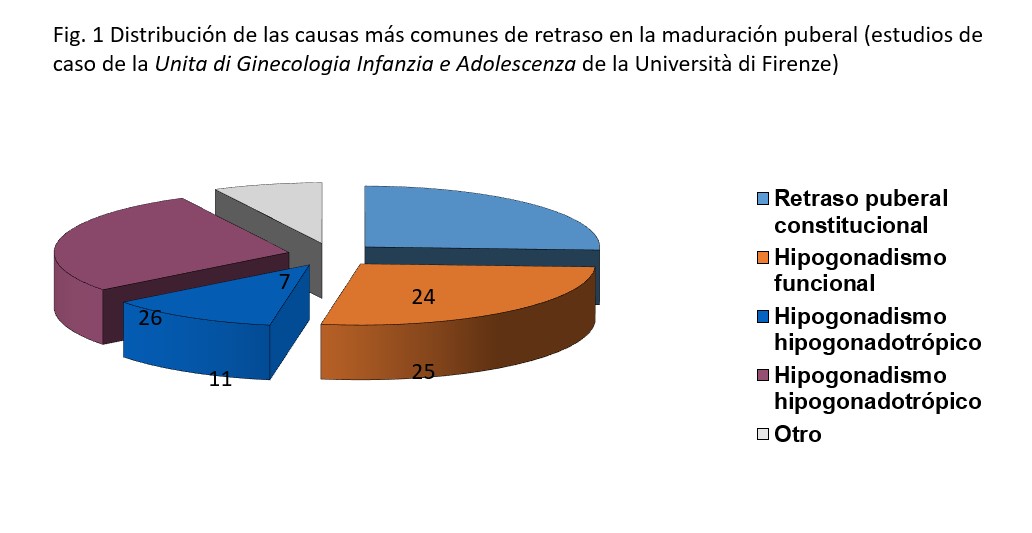
Figura 1: Distribución de las causas más comunes de retraso en la maduración puberal (estudios de caso de la Unita di Ginecologia Infanzia e Adolescenza de la Università di Firenze)
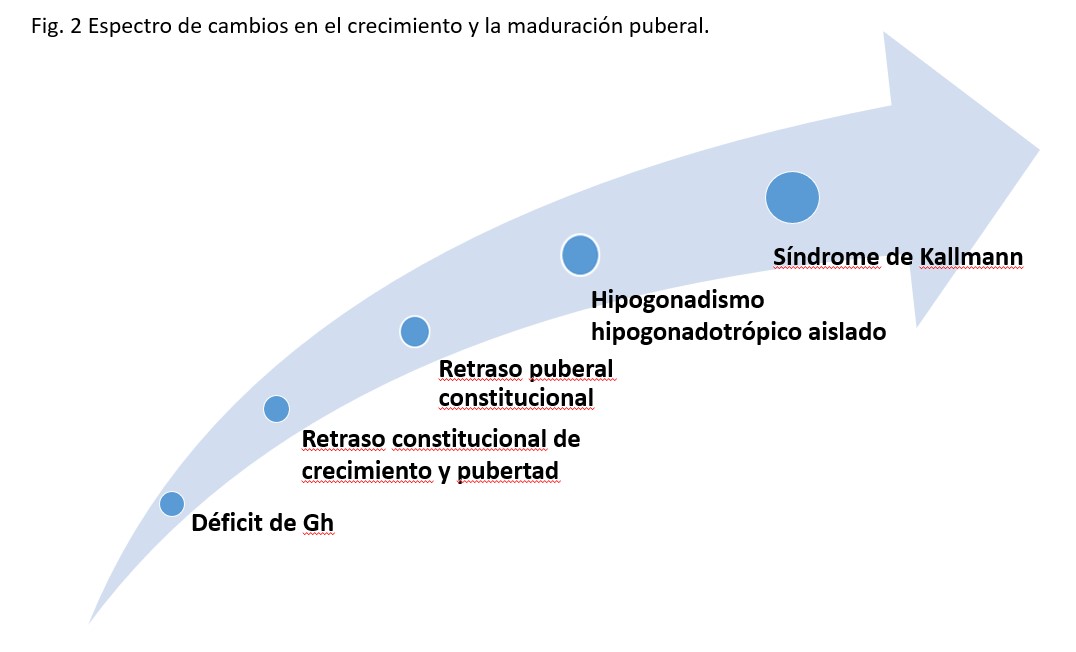
Figura 2: Espectro de cambios en el crecimiento y la maduración puberal.
Referencias Bibliográficas:
[1] O'Connor SM, Culbert KM, Mayhall LA y col. Differences in genetic and environmental influences on body weight and shape concerns across pubertal development in females. J Psychiatr Res 2020; 121:39-46.
[2] Bruni V, Dei M, Morelli C y col. Body composition variables and leptin levels in functional hypothalamic amenorrhea and amenorrhea related to eating disorders. J Pediatr Adolesc Gynecol. 2011; 24(6):347-52.
[3] Pinto Gomes FC, Neto da Cunha M B C, Rocha Marcondes MG y col. Hypopituitarism due to to hydrocephalus: case report and review of the literature. Pediatr Neurosurg 2011; 47(4): 303-6.
[4] Auer MK, Stieg MR, Crispin A y col. Primary Empty Sella syndrome and the prevalence of hormonal dysregulation. Dtsch Arztebl Int 2018; 115(7): 99-105.
[5] Joshi MN, Whitelaw BC, Carroll PV. Hypophysitis: diagnosis and treatment. European Journal Endocrinology 2018; 179, R151–R163.
[6] Gutenberg A, Larsen J, Lupi I, y col. A radiologic score to distinguish autoimmune hypophysitis from nonsecreting pituitary adenoma preoperatively. Am J Neuroradiology 2009; 30: 1766–1772.
[7] Topaloğlu AK. Update on the genetics of Idiopathic Hypogonadotropic Hypogonadism. J Clin Res Pediatr Endocrinol 2017; 9 (Suppl 2):113-122.
[8] Young J, Xu C, Papadakis GE y col. Clinical management of Congenital Hypogonadotropic Hypogonadism. Endocr Rev 2019; 40(2):669-710.
[9] Varimo T, Miettinen PJ, Känsäkoski J y col. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? An analysis of a large patient series from a single tertiary center. Hum Reprod 2017; 32(1):147-153.
[10] Bozzola M, Bozzola E, Montalbano C. Delayed puberty versus hypogonadism: a challenge for the pediatrician. Ann Pediatr Endocrinol Metab 2018; 23(2):57-61.
[11] Bry-Gauillard H, Larrat-Ledoux F, Levaillant JM y col. Anti-Müllerian Hormone and ovarian morphology in women with Isolated Hypogonadotropic Hypogonadism/Kallmann Syndrome: effects of Recombinant Human FSH. J Clin Endocrinol Metab 2017; 102(4):1102-1111.
[12] Mosbah H, Bouvattier C, Maione L y col. GnRH stimulation testing and serum inhibin B in males: insufficient specificity for discriminating between congenital hypogonadotropic hypogonadism from constitutional delay of growth and puberty. Hum Reprod. 2020; 35(10):2312-2322.
[13]Wong A P, Pipitone J, Park MTM y col. Estimating volumes of the pituitary gland from T1-weighted magnetic-resonance images: Effects of age, puberty, testosterone, and estradiol. Neuroimage 2014; 94: 216-221.
[14] Whittle S, Barendse M, Pozzi E. Pubertal hormones predict sex-specific trajectories of pituitary gland volume during the transition from childhood to adolescence. NeuroImage 2020; 204: 116256.
[15] Winter S, Ousidhoum A, McElreavey K. Constitutional delay of puberty: presentation and inheritance pattern in 48 familial cases. BMC Pediatr 2016; 16:37-45.
[16] Maione L, Dwyer AA, Bruno Francou B y col. Genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol 2018; 178, R55–R80.
[17] Howard SR, Dunkel L. The genetic basis of delayed puberty. Neuroendocrinology 2018;106: 283–291.
[18] Harrington J, Palmert M. Clinical review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab 2012; 97(9):3056-67.
[19] Raivio T, Miettinen PJ. Constitutional delay of puberty versus congenital hypogonadotropic hypogonadism: Genetics, management and updates. Best Practice & Research Clinical Endocrinology & Metabolism 2019; 33:101316.
[20] Chan YM, Lippincott MF, Kusa TO y col. Divergent responses to kisspeptin in children with delayed puberty. J C I Insight 2018; 3:e99109.
[21] Sari S, Sari E, Akgun V y col. Measures of pituitary gland and stalk: from neonate to adolescence. J Pediatr Endocr Met 2014; 27(11-12): 1071–1076.
[22] Boehm U, Bouloux PM, Dattani MT y col. European Consensus Statement on congenital hypogonadotropic hypogonadism—pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015; 11(9):547-64.
[23] Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346(1-2):21-8.
[24] de Roux N, Young J, Brailly-Tabard S y col. The same molecular defects of the gonadotropin-releasing hormone receptor determine a variable degree of hypogonadism in affected kindred. J Clin Endocrinol Metab 1999; 84(2):567-72.
[25] Hietamäki J, Hero M, Holopainen E. GnRH receptor gene mutations in adolescents and young adults presenting with signs of partial gonadotropin deficiency. PLoS One 2017; 12(11):e0188750.
[26] Kottler ML, Counis R, Bouchard P. Mutations of the GnRH receptor gene: a new cause of autosomal-recessive hypogonadotropic hypogonadism. Arch Med Res. 1999; 30(6):481-5.
[27] Kelly WM, Kucharczykc W, JKucharczyk J y col. Posterior pituitary ectopia: an MR feature of pituitary dwarfism. Am J Neuroradiol 1988; 9(3):453-60.
[28] Bozzola M, Adamsbaum C, Biscaldi I y col. Role of magnetic resonance imaging in the diagnosis and prognosis of growth hormone deficiency. Clin Endocrinol (Oxf) 1996; 45(1):21-6.
[29] Simon D, Hadjiathanasiou C, Garel C y col. Phenotypic variability in children with growth hormone deficiency associated with posterior pituitary ectopia. Clin Endocrinol (Oxf). 2006; 64(4):416-22.
[30] Ladjouze A, Soskin S, Garel C y col. GH deficiency with central precocious puberty: a new rare disorder associated with a developmental defect of the hypothalamic-pituitary area. Eur J Endocrinol 2007; 156(4):463-9.
[31] Murray PG, Hague C, Fafoula O. Associations with multiple pituitary hormone deficiency in patients with an ectopic posterior pituitary gland. Clin Endocrinol (Oxf) 2008; 69(4):597-602.
[32] Martin DD, Wit JM, Hochberg Z y col. The Use of Bone Age in Clinical Practice – Part 1. Horm Res Paediatr 2011; 76: 1–9.
[33] Zhu J, Choa RE, Guo MH y col. A shared genetic basis for self-limited delayed puberty and idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2015; 100(4):E646-54.
[34] Cassatella D, Howard SR, Acierno JS y col. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. European Journal of Endocrinology 2018; 178, 377–388.
[35] Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L y col. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab 2012; 97, E136–E144.
[36] Della Valle E, Vezzani S, Rochira V y col. Prevalence of olfactory and other developmental anomalies in patients with central hypogonadotropic hypogonadism. Frontiers in Endocrinology 2013; 4 (70): 2-5.
[37] Shaw ND, Seminara SB, Welt CK y col. Expanding the phenotype and genotype of female GnRH deficiency. J Clin Endocrinol Metab 2011; 96(3):E566-76.
[38] Tang R, Chen R, Ma M. Clinical characteristics of 138 Chinese female patients with idiopathic hypogonadotropic hypogonadism. Endocr Connect 2017; 6(8):800-810.
[39] Raivio T, Falardeau J, Dwyer RS y col. Reversal of Idiopathic Hypogonadotropic Hypogonadism. N Engl J Med 2007; 357 (9): 863-73.
[40] Quinton R, Cheow HK, Tymms DJ y col. Kallmann’s syndrome: is it always for life? Clin Endocrinol (Oxf) 1999; 50: 481–5.
[41] Laitinen EM, Tommiska J, Sane T y col. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One 2012; 7: e39450.
[42] Sidhoum VF, Chan YM, Lippincott MF y col. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab 2014; 99: 861–70.
[43] Mao JF, Xu HL, Duan J, y col. Reversal of idiopathic hypogonadotropic hypogonadism: a cohort study in Chinese patients. Asian J Androl 2015; 17(3):497-502.
[44] Dewailly D, Boucher A, Decanter C y col. Spontaneous pregnancy in a patient who was homozygous for the Q106R mutation in the gonadotropin-releasing hormone receptor gene. Fertil Steril 2002; 77: 1288–1291
[45] Gianetti E, Tusset C, Sekoni D N y col. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab 2010; 95(6):2857-67.
[46] Matthews D, Bath L, Högler W y col. Hormone supplementation for pubertal induction in girls. Arch Dis Child 2017; 102: 975e80.
[47] Ankarberg-Lindgren, C., Kristrom, B. & Norjavaara, E. Physiological estrogen replacement therapy for puberty induction in girls: a clinical observational study. Horm. Res. Paediatr 2014; 81, 239 –244.
[48] Ankarberg-Lindgren C, Aneta Gawlik A, Kriström B y col. Estradiol matrix patches for pubertal induction: stability of cut pieces at different temperatures. Endocrine Connections 2019;8: 360 –366.
[49] Chan Y, Feld A, Jonsdottir-Lewis E. Effects of the timing of sex-steroid exposure in adolescence on adult health outcomes. J Clin Endocrinol Metab 2019; 104(10):4578-4586.
(¹) Ginecóloga Endocrinóloga profesional independiente en Florencia (Italia)
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
(²) Doctora en formación en la Azienda Ospedaliera Universitaria Careggi de Florencia (Italia)
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.