

Amenorrea primaria: cuando la genética nos interpela
Primary amenorrhea: when genetics challenges us
Caso Clínico
Dres. Sayago Pablo Ariel,¹ Martínez María Alejandra,² Ercoli Gabriel.³
Resumen
Se define como amenorrea primaria a la ausencia de menstruación a los 16 años, independientemente del crecimiento y la presencia o ausencia de caracteres sexuales secundarios. Dentro de las amenorreas hipogonadotróficas congénitas, encontramos al síndrome de Kallmann, un cuadro caracterizado por la asociación de amenorrea primaria y anosmia. Se presenta un caso con este diagnóstico presuntivo. En el examen clínico, se constató Estadio III de Tanner-Marshall para desarrollo mamario y vello pubiano. Se comprobó permeabilidad vaginal. En olfatometría, se constató anosmia. En la resonancia magnética de cerebro, se diagnosticó aracnoidocele. El resultado del estudio citogenético fue: 45,X[6]/ 46,XX[44]. Se sugirió efectuar un estudio de exoma clínico dirigido a genes asociados a cuadros de hipogonadismo hipogonadotrófico con anosmia. En la evaluación clínica de una adolescente que consulta por amenorrea, no se debe obviar la evaluación clínica del olfato; asimismo, indagar sobre antecedentes familiares de infertilidad y/o alteraciones del olfato, sobre todo en varones.
Palabras clave: amenorrea, anosmia, Kallmann, aracnoidocele, monosomía del X.
Summary
Primary amenorrhea is defined as the absence of menstruation at age 16 regardless of growth and the presence or absence of secondary sexual characteristics. Within congenital hypogonadotrophic amenorrhea, we find Kallmann syndrome, a condition characterized by the association of primary amenorrhea and anosmia. A case is presented with this presumptive diagnosis. The clinical examination revealed Tanner-Marshall Stage III for breast development and pubic hair. Vaginal patency is checked. In olfactometry, anosmia is confirmed. Arachnoidocele is diagnosed in magnétic resonance imaging of the brain. The result of the cytognetic study is: 45,X [6]/ 46,XX[44]. It was suggested to carry out a clinical exome study aimed at genes associated with hypogonadism is awaited. hypogonadotrophic with anosmia. In the clinical evaluation of an adolescent who consults for amenorrhea, we should not ignore the clinical evaluation of smell, and inquire about a family history of infertility and/or smell disorders, especially in men.
Keywords: amenorrhea, anosmia, Kallmann, arachnoidocele, X monosomy.
Introducción
La amenorrea no es una entidad clínica, sino sólo un síntoma que puede observarse en distintas afecciones genitales y extragenitales.1. Se define como amenorrea primaria a la ausencia de menstruación a los 16 años independientemente del crecimiento y la presencia o ausencia de caracteres sexuales secundarios. Sus causas más frecuentes son la disgenesia gonadal en un 50% de los casos y las del eje hipotálamo-hipófiso-gonadal en un 25%, mientras que el resto responde a otras causas, entre las que se encuentran las malformaciones útero-vaginales.2 En Argentina, la edad media de la menarca es a los 12,6 años y el 97% de las niñas presentan desarrollo de caracteres sexuales secundarios a los 13 años de edad.2
La presencia o no de caracteres sexuales secundarios y los valores de gonadotrofinas orientan en el tipo de amenorrea y su origen. Así, la falta de desarrollo de los caracteres sexuales secundarios sugiere una causa central congénita o adquirida y si el mismo está presente, se piensa en una causa genital.
Según el nivel de gonadotrofinas se las puede clasificar en: hipogonadotróficas, hipergonadotróficas y normogonadotróficas, y según el nivel de estradiol en hipoestrogénicas, hiperestrogénicas y normoestrogénicas.2
El término anosmia define la pérdida total de la capacidad olfativa. Puede clasificarse en anosmia de transmisión cuando es provocada por una obstrucción a nivel nasal, o neurosensorial cuando la lesión se localiza en cualquier punto de la vía nerviosa entre el epitelio olfativo y la corteza cerebral.3
Dentro de las amenorreas hipogonadotróficas congénitas se encuentra al Síndrome olfato-genital o Síndrome de Kallmann, un cuadro que se caracteriza por la asociación de amenorrea primaria y anosmia. Hasta la fecha, diferentes genes que afectan el desarrollo/migración de las neuronas GnRH han sido implicados en este síndrome, pero el conocimiento de su base genética sigue siendo incompleto.4
La escasa frecuencia de este síndrome, y algunos hallazgos que surgieron en el estudio y seguimiento de este caso en particular, motivan a los autores a comunicar el mismo.
Caso clínico
L., de 18 años, consulta a principios de 2021 derivada desde una institución particular por ausencia de menstruación asociada a falta de percepción de olores de larga data. Cabe mencionar que ya había consultado en 2020 por dicho motivo, pero discontinuó consultas debido al aislamiento social preventivo y obligatorio dispuesto al inicio de la pandemia provocada por Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV 2).
Como antecedentes personales, se rescata bronquiolitis a los tres meses de edad, quemadura accidental en brazo izquierdo a los cuatro años y también un “fuerte” golpe en la cabeza por caída a los 5-6 años. Se descartó maltrato infantil; posterior a ello, refiere sólo percibir olores fuertes como el alcohol y acetonas.
Dentro de los antecedentes heredofamiliares, se destaca padre fallecido en 2002 debido a un tumor cerebral. Los antecedentes ginecológicos son telarca y pubarca referidas a los 11 años. No ha presentado menarca. No había iniciado relaciones sexuales. En cuanto a familia-actividad actual y hábitos, convive con su madre y una hermana mayor, es estudiante universitaria y refiere realizar caminatas.
En el examen físico la tensión arterial es de 100-60 mm Hg, el peso es de 68.900 Kg (p90-97) y la talla de 1.63 cm (p50-75). IMC: 26.
No presentó hirsutismo y se detectó palpación tiroidea normal. Las mamas, sin secreción por pezón, Estadio III de Tanner-Marshall. Vello en pubis: Estadio III de Tanner-Marshall. La palpación abdominal y los genitales externos no mostraron particularidades. Se comprobó permeabilidad vaginal con hisopo.
Se realizó test de embarazo (negativo) y test de reacción en cadena de la polimerasa (PCR) para SARS CoV 2 (negativo) ( estudio solicitado dado que la consulta transcurre en pandemia). Se solicitó ecografía ginecológica, laboratorio completo con perfil hormonal y estudio genético. Asimismo, se realizó prueba de progesterona en dos oportunidades: ambas negativas. Posteriormente, se efectuó prueba de estrógenos-progesterona: positiva.
La ecografía ginecológica mostró el útero en AVF de forma, tamaño y ecoestructura conservada; diámetro longitudinal de 64mm, diámetro transverso de 14mm y diámetro anteroposterior de 48mm; el endometrio lineal midió 1,9 mm. El ovario derecho midió 18x11 mm y el ovario izquierdo, 8x13 mm. El fondo de saco de Douglas estaba libre.
El laboratorio hormonal mostró: FSH 0,6 mUI/ml (0,9-11) - LH 0,1 mUI/ml (0,8-12) - estradiol 33 pg/ml (25-60) - PRL 7.7 ng (2,8-29,9) - TSH 2,66 uUI/ml (0,4-4,8) - DHEA 3,9 ng/ml (1,4-18) - S-DHEA 694 ng/ml (800-3600 ng/ml) - Testosterona total 0,16 ng/ml (0,3-0,9) - Testosterona libre 4,4 pg/ml - 17OH Progesterona 0,6 ng/ml (0,4-1,5) – A4 0,8 ng/ml (0,5-3,7) - Hormona Antimulleriana 2,69 ng/ml (prepúberes hasta 10 -Adultos 0,2 a 5,0 - posmenopáusicas hasta 0,14 ng/ml) - ACTH 14,2 pg/ml (hasta 46); Cortisol 13,1 pg/ml (4,3-22,4) - Calcio total 9,7 ng/dl (8,5-10.5) - Calcio iónico 4,9 ng/dl (4,25-5,25) - Mg+ 2,2 ng/dl (1,7-2,5) - Fosfatemia 4,40 mg/dl (2,5-5,6) - PTH 17,4 pg/ml (14-77); Vitamina D3 26,20 ng/ml (Deficiencia menor a 20, Insuficiencia: 21-29; Suficiencia: mayor a 30 ng/ml).
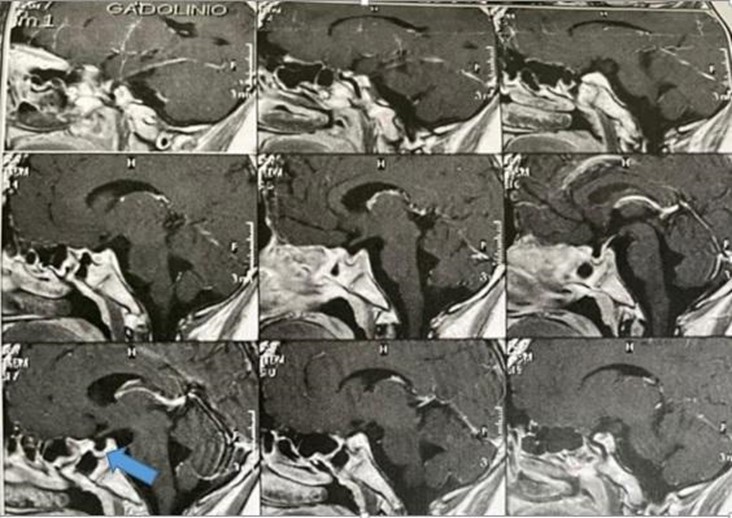
La resonancia magnética (RM) de encéfalo informó que la hipófisis presentaba reducción de sus diámetros en sentido cráneo caudal. También evidenciaba aracnoidocele intraselar, asociado a síndrome de silla turca vacía (Gráfico 1).
Con diagnóstico presuntivo de Síndrome de Kallmann, se decidió iniciar tratamiento con 17 B estradiol 2 mg/día, con el plan de añadir progesterona natural micronizada 200 mg del día 16 al 26º del ciclo, vía oral.
Se solicitó seguimiento conjunto con el Servicio de Endocrinología, al cual se le pidió olfatometría. Informó anosmia.
Se realizó estudio genético en cultivo de sangre periférica con técnica de Bandeo G (GTW) con nivel de resolución 400-450 bandas; se analizaron 50 metafases y no se observaron anomalías cromosómicas estructurales. Se informaron dos líneas celulares, una de 45 cromosomas en ausencia de un cromosoma X (8%) y otra de 46 cromosomas (92%): 45,X[6]/ 46,XX[44].
Como estudios adicionales se sugirió realizar exoma clínico, ecocardiograma y audiometría ambos resultaron normales. La densitometría mineral ósea (DMO) informó Z-score: columna lumbar: -2,7. Fémur izquierdo: -1,5.
Actualmente, se encuentra en seguimiento, ciclando mensualmente y aguardando resultado de exoma clínico, recientemente efectuado, ante sospecha de Síndrome de Kallmann.
La ecografía ginecológica luego de tres meses de iniciado el tratamiento informó: útero en AVF de forma, tamaño y ecoestructura conservada, diámetro longitudinal de 70mm, diámetro anteroposterior de 22mm y el endometrio de 4 mm. El ovario derecho midió 23mm x 8mm x 21mm (vol. 4,14mm) y el ovario izquierdo, 24 x 13 x 22 (vol. 4,08mm). Fondo de saco de Douglas, libre.
Discusión y comentarios
El término silla turca vacía se refiere a una condición anatómica causada por una hernia intraselar del espacio subaracnoideo dentro de la silla turca a través de un diafragma selar incompetente. El tejido hipofisario rechazado y comprimido puede ser normal o tener diferentes grados de afectación funcional. Algunos autores consideran que el hallazgo imagenológico de una silla turca vacía en individuos asintomáticos no constituye propiamente un síndrome, sino una variante anatómica. El término síndrome de la silla turca vacía lo reservan para los pacientes con manifestaciones clínicas o funcionales. En general, la mayor parte de los autores no diferencia ambas situaciones.5
El término aracnoidocele es menos utilizado, pero es más apropiado, pues la silla turca en realidad no está vacía, sino ocupada por líquido cefalorraquídeo y la hipófisis, que, rechazada y comprimida, se encuentran en su interior. Se ha calculado que el 22% de la población general puede tener un defecto del cierre del diafragma selar. El hallazgo de una hipófisis que no ocupa toda la silla turca es una observación común, incluso en los individuos normales.5,6. Las deficiencias hormonales son menos del 20% de los casos y la afectación del eje somatotrófico es la más frecuente, seguida por el hipogonadismo hipogonadotrófico.7
Evolutivamente, el olfato, considerado el más primitivo de los sentidos, continúa siendo uno de los menos conocidos y menos estudiados. La patología del olfato puede alterar de manera significativa la calidad de vida del paciente. 3
En 2004, los investigadores estadounidenses Linda Buck y Richard Axel recibieron el Premio Nobel de Medicina y Fisiología por sus estudios en fisiología olfatoria. Los científicos demostraron que los receptores olfatorios forman parte de una amplia familia de receptores acoplados a las proteínas G.3
Las anosmias neurosensoriales se desarrollan cuando la lesión se localiza en cualquier punto de la vía nerviosa entre el epitelio olfativo y la corteza cerebral. Por lo general, los pacientes no dimensionan la pérdida del olfato hasta que son formalmente testeados.3 Dentro de los mecanismos neuroinvasivos, se sabe que, además de la vía hematógena, los virus pueden infectar neuronas periféricas y migrar a través de ellas hasta el sistema nervioso central. La vía transneuronal a través de la vía olfatoria podría relacionarse con la temprana afectación en muchos pacientes en forma de anosmia, síntoma reconocido como biomarcador clínico precoz de la infección por COVID-19 que puede preceder al cuadro clínico completo o presentarse aisladamente en formas leves.8
Se ha detectado ARNm de SARS-CoV-2 dentro de la glándula hipófisis, con efectos citopáticos y reducción del número de células somatotropas, tirotropas y corticotropas y de la tinción de inmunoreactividad para GH, TSH y ACTH.9
La expresión de la enzima convertidora de angiotensina 2 (ACE2), receptor celular para SARS-CoV-2, en el hipotálamo fue confirmada por Chigr et al, quienes identificaron su presencia en el núcleo paraventricular. El genoma del SARS-CoV-2 se ha detectado en líquido cefalorraquídeo, lo que confirma que este virus puede afectar cualquier parte del cerebro, al atravesar la barrera hematoencefálica, incluidos el hipotálamo y la hipófisis.9-10
Sin embargo, no se ha hallado la expresión de ACE2 en neuronas del bulbo olfatorio por lo tanto la anosmia sugiere daño al receptor olfativo.8 Las estrategias de mitigación y control del COVID-19, así como los confinamientos y el distanciamiento social, han provocado aumento del estrés psicológico, la depresión y la ansiedad. El estrés psicológico es un factor de riesgo conocido para el hipogonadismo hipotalámico. También cualquier enfermedad grave, incluida el COVID 19 puede provocar amenorrea o menstruaciones poco frecuentes lo que estaría relacionado con la liberación disrítmica de GnRH. Este mecanismo de protección permite el desvío de recursos energéticos de la reproducción a la respuesta inmunitaria.9-11
La deficiencia aislada de hormona liberadora de gonadotrofina constituye, en realidad, un amplio espectro clínico que incluye a una variedad de trastornos incluido el Síndrome de Kallmann, su mayor componente, es decir, hipogonadismo hipogonadotrófico con anosmia por un lado y, por otro, al hipogonadismo hipogonadotrófico idiopático normósmico; estos representan los extremos más graves del espectro. Además de estos trastornos se incluye la amenorrea hipotalámica funcional, el retraso constitucional de la pubertad y al hipogonadismo hipogonadotrófico de inicio en la edad adulta. Una gran superposición clínica y genética caracteriza el espectro de la deficiencia aislada de hormona liberadora de gonadotrofina. 12
La ocurrencia del Síndrome de Kallmann puede responder a un patrón autosómico dominante, autosómico recesivo o ligado al cromosoma X. Se estima que el 60% de los casos son esporádicos. Las afectadas presentan constitución cromomosómica XX.12
Algunos de los genes más importantes son: KAL1 (Xp22.31); ANOS1(Xp22.31) ambos recesivos ligados al cromosoma X. Autosómicos dominantes son: FGFR(8P11.23), FGF8(10q24.32), SOX10(22q13.1) y GNRH(8p21.2) con carácter recesivo.13,14,15 Normalmente, durante la embriogénesis, las células inmaduras de GnRH migran desde el epitelio olfatorio a través de la placa cribiforme del etmoides, hacia el bulbo olfatorio en desarrollo. Los defectos en el desarrollo de estas neuronas o en su función secretora dan como resultado la deficiencia aislada de GnRH.12
La frecuencia en hombres es de 1:10.000 y en mujeres 1:50.000; por lo tanto, los hombres pueden verse más afectados en una proporción 5:1 en casos esporádicos y de 2:1 en casos familiares.16 (Tabla N 1). En ambos sexos, se constatan niveles extremadamente bajos de gonadotrofinas y esteroides gonadales, durante el período de “minipubertad”. Se asocian anomalías congénitas cardíacas, discapacidad auditiva, labio o paladar hendido, agenesia renal unilateral, polidactilia, sindactilia, sincinesias, epilepsia, ataxia cerebelosa.17-18
El diagnóstico diferencial debe realizarse con tumores que causan hipogonadismo hipogonadotrófico adquirido, trastorno constitucional del desarrollo, amenorrea hipotalámica, hipogonadismo hipogonadotrófico de inicio en la edad adulta.12
Se ha descrito que hasta un 20% pueden experimentar en algún momento de la vida adulta una recuperación al estado reproductivo, es decir. una inversión del hipogonadismo, luego de discontinuar el tratamiento hormonal, aunque es posible que esto no se mantenga a largo plazo. 4,12,17,18
Esta paciente presentó amenorrea primaria con desarrollo de caracteres sexuales secundarios, se descartaron malformaciones útero-vaginales por examen físico y ecografía. Además, sus gonadotrofinas se encontraban disminuidas, las pruebas de progesterona negativas indicaron que el endometrio no ha sido “cebado” suficientemente por estrógenos, el resto del perfil hormonal fue normal.
La RM de cerebro diagnosticó un aracnoidocele. Los autores consideran improbable que la instalación del aracnoidocele haya causado la deficiencia aislada de gonadotrofinas, ya que, como menciona la literatura, puede no haber necesariamente síntomas de hipofunción. Por otra parte, ésta imagen no explicaría la anosmia. En nuestro caso, tal síntoma en contexto de pandemia, obligó a la investigación de SARS-CoV2 cuyo resultado fue negativo.
El estudio citogenético informó la presencia de un mosaicismo cromosómico, consistente en la presencia de dos poblaciones celulares: una línea celular minoritaria con monosomía del cromosoma X y otra línea celular mayoritaria femenina normal, con un número modal de 46 cromosomas con complemento sexual XX. Cabe destacar que, a partir de un cariotipo con bandeo G en sangre periférica, no es posible conocer cuál es el porcentaje real de la monosomía del X a nivel gonadal y su implicancia a nivel reproductivo. Por otro lado, ésta suele cursar con hipogonadismo hipergonadotrófico mientras que la paciente presenta un hipogonadismo hipogonadotrófico con anosmia, sin otros hallazgos compatibles con síndrome de Turner, lo que orientó a pensar en un Síndrome de Kallmann.
El estudio del exoma clínico dirigido a genes asociados a cuadros de hipogonadismo hipogonadotrófico con anosmia, se refiere a un enfoque de secuenciación masiva genética, por técnica Next Generation Sequencing, que se centra en analizar específicamente las regiones codificantes de los genes relacionados con el desarrollo de hipogonadismo hipogonadotrófico y anosmia. Se busca identificar posibles variantes patogénicas que puedan estar implicadas en la causa subyacente de esta condición, permitiendo así un diagnóstico más preciso y personalizado, lo que brindará claridad sobre su patrón de herencia y su pronóstico. De este modo será de utilidad para completar el asesoramiento genético personal y familiar. 19
La DMO siempre la solicitamos cuando tenemos una amenorrea mayor a 6 meses, se mide el Z-score en referencia a población especifica ajustada por edad, sexo y etnia. Este caso presentó un rango de osteoporosis.
En la evaluación clínica de una adolescente que consulta por amenorrea primaria, no debe obviarse la evaluación clínica del olfato, ya que las alteraciones cuantitativas de este pueden pasar desapercibidas. Asimismo, es importante indagar sobre antecedentes familiares por ejemplo de infertilidad y/o alteraciones del olfato, sobre todo en varones.
La relación entre sexo y olfato encuentra aún hoy muchos interrogantes por resolver. Nos muestra que el delicado y complejo desarrollo del sistema endocrinológico durante la temprana vida embrionaria gravita sobre manifestaciones clínicas más tardías, en etapa adolescente o adulta.
Tabla 1. Características clínicas del Síndrome de Kallmann en ambos sexos, según períodos.
|
|
Período neonatal e infancia |
Adolescencia |
Adulto |
|
Varón |
Criptorquidia y micropene con longitud menor a 2,5 cm. |
Virilización parcial. Líbido baja. Volumen testicular prepuberal (menor a 4ml), disminución de la masa muscular. Hábito corporal eunucoide. |
Infertilidad. Aumento de senos o falta de barba con consiguientes problemas psicológicos. |
|
Mujer |
Sin signos clínicos específicos. |
Desarrollo parcial o ausencia de mama. Empuje puberal atenuado. Amenorrea primaria. |
Infertilidad. Fracturas osteoporóticas. |
Cuadro de elaboración propia basado en: Dodé C, Hardelin J. “Kallmann Syndrome.” European Journal of Human Genetics ,17( 2008);139-146.https://doi.org/10.038/ejh.2008.206. 17
Liu Y, Zhi X. “Advances in genetic diagnosis of Kallmann Syndrome and genetic interruption”. Reproductive Sciences. Vol 29.(2022): 1697-1709. https://doi.org/10.007/543032-021-00638-8 .18
Gráfico 1. RM cerebral con contraste de gadolinio con foco en silla turca (imagen ampliada). Se señala con flecha imagen de aracnoidocele.
Bibliografía
- Calatroni C, Ruiz V; Tratamiento de las amenorreas; En: Calatroni C, Ruiz V. Terapéutica ginecológica; capítulo 13; Bs As; Editorial Médica Panamericana; 1988; 333.
- Sociedad Argentina de Endocrinología Ginecológica y Reproductiva. SAEGRE; Amenorrea primaria. En: Sociedad Argentina de Endocrinología Ginecológica y Reproductiva. Manual clínico de endocrinología ginecológica y reproductiva; Capítulo 8; Bs As; Ediciones Journal;2020;139.
- Carrillo B, Carrillo V, Asinga A et al. “Diagnóstico en la patología del olfato: Revisión de la literatura”. Revista de Otorrinolaringología y Cirugía de Cabeza y Cuello. Vol 77. (2017): 531-560.
- Swee DS, Quirton R and Maggi R. “Recents advances in understanding and managing Kallmann Syndrome”. Faculty Reviews.10(2021):10-37. https://doi.org/10.12703/r/10-37
- Hung Llamos S; Silla turca vacía. En: Hung Llamos S. Endocrinología en Ginecología. Tomo 1. Cap. 3. La Habana. Editorial Cs Médicas;2006;93-94.
- Delgado-Hernández A, Verduzco-Mendoza A. Luna Reyes F. “Análisis de la probabilidad conjunta y a posteriori entre aracnoidocele selar primario, sus comorbilidades y enfermedad audiovestibular”. Cirugía y Cirujanos. 83. (2015): 459-466. http:// dx.doi.org/10.016/j.circir.2015.04.031
- García Soto J. Hipofunción hipofisaria; En: Dorantes Cuellar A, Martinez C, Ulloa Aguirre A. Endocrinología Clínica de Dorantes y Martínez; Capítulo 6, México; Sociedad Mexicana de Nutrición y Endocrinología; El Manual Moderno;2016; 64.
- Martínez Hernández E, Velasco F; Mecanismos fisiopatogénicos de afectación neurológica; En: Manual COVID 19 para el neurólogo general. Capítulo 4. Madrid. Ediciones SEN. Sociedad Española de Neurología; 2020; 25-29.
- Mirza S, Sheik A, Barbera Michaela et al. “Covid 19 and the endocrine system: A review of the current information and misinformation”. Infect. Disp.Resp. 2022, 14(2):184-197. https://doi.org/10.3390/idr14020023
- Clarke S, Abbara A, Waljit S. “Impact of covid 19 in the the endocrine system. Mini-Review” Endocrinology. Vol 163. N 1.(2022):1-19. https://doi.org/10.1210/Endocr/bqab203.
- Sharp G, Faser A, Sawyer G et al. “The covid-19 pandemic and the menstrual cycle: research gaps and opportunities”. International Journal of Epidemiology. Vol 51. N 3 (2022):691-700. https://doi.org/10.1093/ije/dyab239
- Stamou M, Georgopoulus N. “Kallmann syndrome: phenotype and genotype of hipogonadotropic hipogonadism”. Metabolism; 86:(2018):124-134. https://doi.org/10.1016/metabol.2017.10.01
- Balasubramanian R, Crowley W Jr. Isolated Gonadotropin-Releasing Hormone (GnRH) Deficiency. 2007 May 23 [updated 2022 May 12]. In: Adam M, Feldman J, Mirzaa G, Pagon R, Wallace S, Bean L, Gripp K, Amemiya A, Editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. PMID: 20301509. Bookshelf ID: NBK1334.Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1334/ Consultado el 07/03/2024.
- Patil VA, Lila AR, Shah N et al.” Genetic spectrum of Kallmann syndrome: Single-center experience and systematic review”. Clin Endocrinol (Oxf). 97-6 (2022): 804-813. doi: 10.1111/cen.14822. Epub 2022 Sep 30. PMID: 36138264.
- Patil VA, Lila AR, Shah N et al.(2022). “Genetics of Kallmann syndrome: Single-center Experience with Review of NGS based Genetic Studies”. Research Square (2022):1-10. doi: 10.21203/rs.3.rs-1648832/v1.
- Bueno Prado A, Rodríguez Sousa L, Lessa Junior L et al. “Síndrome de Kallmann”. Brazilian Journal of Health Review. Vol 5.Num 5 (2022): 18600-18608. https://doi.org:10.34119/bjhrv5n5-067
- Dodé C, Hardelin J. “Kallmann Syndrome.” European Journal of Human Genetics,17 ( 2008); 139-146. https://doi.org/10.038/ejh.2008.206
- Liu Y, Zhi X. “Advances in genetic diagnosis of Kallmann Syndrome and genetic interruption”. Reproductive Sciences. Vol 29.(2022): 1697-1709. https://doi.org/10.007/543032-021-00638-8
- Shickh S, Mighton C, Uleryk E et al. “The clinical utility of exome and genome sequencing across clinical indications: a systematic review”. Hum Genet.140-10 (2021):1403-1416. doi: 10.1007/s00439-021-02331-x.
- Médico de planta especialista en Ginecología y Obstetricia. Servicio de Tocoginecología del Hospital Zonal Gral. de Agudos Narciso López de Lanús.
Gral. Guido, B1824 Lanús, Provincia de Buenos Aires - Médica de planta especialista en Ginecología y Obstetricia. Servicio de Ginecología del Hospital Gral. de Agudos Bernardino Rivadavia.
Av. Las Heras 2670. Ciudad Autónoma de Buenos Aires. - Médico especialista en Genética Médica. Director Médico de Gempre: Genómica y Medicina Preventiva de Precisión.
Viamonte 1621 9no piso, C1019 Cdad. Autónoma de Buenos Aires